File:Phylogenetic tree of coronaviruses.jpg
No higher resolution available.
Phylogenetic_tree_of_coronaviruses.jpg (571 × 451 pixels, file size: 75 KB, MIME type: image/jpeg)
Captions
Captions
Coronavirus
Summary
edit{kind=link}
| Description |
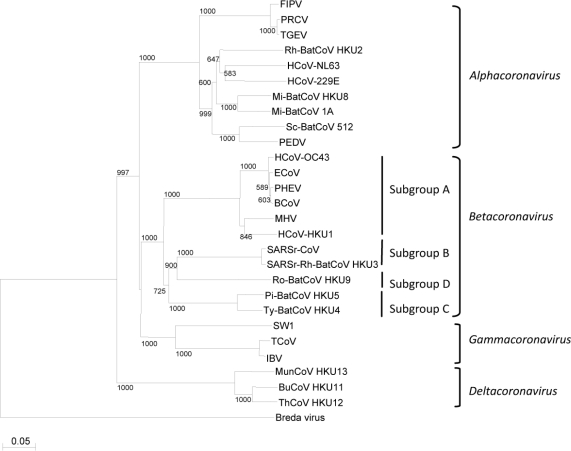
English: Phylogenetic analysis of RNA-dependent RNA polymerases (Pol) of coronaviruses with complete genome sequences available. The tree was constructed by the neighbor-joining method and rooted using Breda virus polyprotein. Bootstrap values were calculated from 1000 trees. 1118 amino acid positions in Pol were included. The scale bar indicates the estimated number of substitutions per 20 amino acids. All abbreviations for the coronaviruses were the same as those in Figure 1. |
| Date | |
| Source | https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3185738/ |
| Author | Patrick C. Y. Woo, Yi Huang, Susanna K. P. Lau, and Kwok-Yung Yuen |
Licensing
edit{kind=link}
This file is licensed under the Creative Commons Attribution 3.0 Unported license.
- You are free:
- to share – to copy, distribute and transmit the work
- to remix – to adapt the work
- Under the following conditions:
- attribution – You must give appropriate credit, provide a link to the license, and indicate if changes were made. You may do so in any reasonable manner, but not in any way that suggests the licensor endorses you or your use.
File history
Click on a date/time to view the file as it appeared at that time.
| Date/Time | Thumbnail | Dimensions | User | Comment | |
|---|---|---|---|---|---|
| current | 02:42, 7 March 2020 | | 571 × 451 (75 KB) | Guest2625 (talk | contribs) | Uploaded a work by Patrick C. Y. Woo, Yi Huang, Susanna K. P. Lau, and Kwok-Yung Yuen from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3185738/ with UploadWizard |
You cannot overwrite this file.
File usage on Commons
There are no pages that use this file.
File usage on other wikis
The following other wikis use this file:
- Usage on bn.wikipedia.org
- Usage on en.wikipedia.org
- Usage on fa.wikipedia.org
- Usage on fr.wikipedia.org
- Usage on ig.wikipedia.org
- Usage on sq.wikipedia.org
- Usage on su.wikipedia.org
{kind=link}